| Home | AmMin | GMR | RiMG | Collectors Corner | Directory | Short Courses | |
|
|
|||||||

|
|
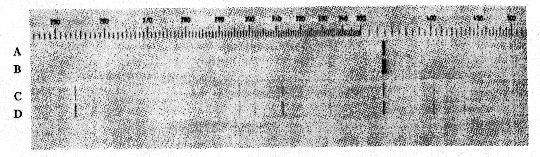
Volume 31, pages 527-538, 1946 K. J. MURATA AND ROBERT L. SMITH.** ABSTRACT The bright red fluorescence of some halite from California dry lake deposits and from a salt spring in Idaho is due to coactivation by minute amounts of manganese and lead. Neither manganese nor lead alone activates the red fluorescence. Chemical, spectrographic, and synthetic experiments, which identify manganese and lead as coactivators and throw light on the mode of precipitation of these elements by growing halite crystals, are described. The marked preferential coprecipitation of lead by growing halite crystals results in zonation of fluorescence in crystals and restriction of fluorescence to the first crops of crystals, when the supply of lead is limited. INTRODUCTION While treating specimens of the natrolite rock from San Benito County, California, with hydrochloric acid in order to free crystals of enclosed neptunite, the junior author noticed that the halite obtained by evaporation of the resulting solution fluoresced red1 under the ultraviolet lamp (Mineralight). This showed that it was feasible to synthesize red-fluorescing salt from water solutions, once the element activating the fluorescence was identified, and further suggested that an explanation might be found for the red fluorescence of halite from two localities in California and from a locality in Idaho. When examined with a small Welch grating spectroscope, the fluorescent light of halite from these localities as well as of the salt crystals that had been accidentally prepared was the same - a continuous spectrum extending from green to red, faint in green and yellow and progressively stronger in orange and red. This spectrum was strikingly similar to that of the well known red-fluorescing calcite from Franklin, New Jersey, for whose fluorescence manganese has been found to be essential.2 A simple experiment soon showed that red-fluorescing halite could be obtained readily from a sodium chloride solution to which a small amount of manganous chloride or sulfate had been added. A spectrographic analysis of the sodium chloride crystals derived from natrolite showed 0.3 per cent of Mn, besides small amounts of other common elements such as magnesium, iron, aluminum, and calcium, and, in addition, a trace of lead. The natural halite samples were also found spectrographically to contain manganese, but in extremely small concentrations. They also contained traces of lead, the significance of which will be discussed later in the paper. PREVIOUS WORK During the course of his extensive study of the effect of radium rays on minerals, Przibram 3 noted that some drusy (secondary?) halite from Stassfurt fluoresced red when exposed to radium rays. Jahoda4 investigated this material further and concluded from results of chemical analyses and synthetic experiments that the red fluorescence was due to small amounts of manganese (0.1 - 0.01% Mn) in the halite. He prepared a manganiferous salt for comparisons with the Stassfurt halite both by fusion methods and by crystallization from water solutions.Oka and Yagi, 5 while studying the effect of various heavy metal ions in promoting the formation of clear sodium chloride crystals from water solutions, noted that crystals obtained from solutions to which small amounts of manganous chloride had been added fluoresced red under ultraviolet light. Randall6 examined a large number of luminescent compounds that are activated by manganese, and described the cathodoluminescence spectrum of a synthetic sample of manganiferous sodium chloride prepared by heating a mixture of sodium chloride and a small amount of a manganese salt to 747°C.The red fluorescence of sodium chloride, therefore, has been related to its content of manganese by several investigators. Our contribution to the subject will consist in proving that our samples from California and Idaho are additional examples of such an activation, and also in describing some new observations on a hitherto unsuspected role that lead plays in this manganese-activated fluorescence of halite. The various aspects of our study will be discussed more or less in the order that they were investigated. This procedure allows us to discuss the different properties of red-fluorescing halite in a logical way, and also serves to emphasize the power of spectrographic methods in the study of fluorescent minerals that are activated by trace elements. LOCALITIES Halite from primary marine rock salt deposits of Carlsbad, New Mexico; Grand County, Utah; Detroit, Michigan, and Retsof, New York, does not fluoresce. Reagent-grade sodium chloride, spectrographically free of manganese but containing traces of lead, is likewise nonfluorescent. The three known American localities that yield red-fluorescing halite are a desert lake deposit near Amboy, California, 7 a similar deposit in Borego Valley, San Diego County, California,8 and the Petersen salt spring9 in the valley of Tygee Creek, Idaho, 3.5 miles west of Auburn, Wyoming.The dry lake salt deposit near Amboy, California, has been briefly described by Phalen. 10 It was mined during World War II to furnish salt for the plant producing magnesium at Las Vegas, Nevada. It would be of great interest to examine the deposit with an ultraviolet lamp at night in order to ascertain the mode of distribution of the red-fluorescing salt. The dry lake deposit of Borego Valley, San Diego County, California, apparently has not yet been described.The brine spring in eastern Idaho, whose salt was found by us to fluoresce red, is believed by Mansfield 11 to owe its origin to circulating ground ground water coming in contact with beds of rock salt in the Jurassic sandstone which underlies the area.It seems likely that halite from other lake deposits and salt springs will be found to be red-fluorescing, just as red-fluorescing calcite has been found in recent years to be of wide occurrence. The specimens from Amboy and Borego Valley, California, are aggregates of anhedral halite crystals with an average diameter of around 5 mm. Some of them are dirty brown in color because of inclusions of clay and organic matter; others are white and translucent. The Idaho samples are efflorescent crusts about 2 cm. thick, made up of poorly crystallized, translucent halite, and tinged a faint red by fine-grained inclusions of iron oxide. WAVELENGTH OF EXCITATION Two types of mercury vapor lamps were first used in our studies. Spectrograms of the light emitted by these lamps, with their respective filters attached, are shown in Fig. 1.
FIG. 1. Spectrograms of light emitted by two types of mercury vapor lamps. Taken with Gaertner medium quartz spectrograph and Eastman I-F plate. A, Inspectolite, 10 secs. B, Same 40 secs. C, Mineralight 20 secs. D, Same, 80 secs. Scale in millimicrons. Spectra A and B are from a high pressure glass mercury vapor lamp (Hanovia Inspectolite) with a power input of 125 watts. Practically all of the light is emitted in a narrow region around 3660 A. U. Spectra C and D are from a low pressure quartz mercury vapor lamp (Ultra-Violet Products Mineralight) with a power input of 14 watts. According to the manufacturer's catalog, this lamp emits about 90% of its total radiant energy through the 2536 A. U. resonance line of mercury. It was found early in our study that both the natural halite specimens and the synthesized manganiferous salt crystals would not fluoresce under the Inspectolite lamp, so all further work was done with the Mineralight lamp. The synthetic crystals stop fluorescing as soon as the lamp is turned off, but the natural samples show varying degrees of whitish phosphorescence which is never very strong and is most marked in some of the specimens from Idaho. It is believed that this phosphorescence is due to another, as yet unidentified activator, and not manganese. SPECTROGRAPHIC ANALYSIS FOR MANGANESE A few typical pieces of red-fluorescing halite from Idaho and from California were analyzed spectrographically for manganese. Each specimen was first dissolved in water, the insoluble impurities filtered off, and the solution taken to dryness, the activator being water soluble. The crystallized salt was ground thoroughly and duplicate 15 mg. portions were used to obtain the spectrograms on Eastman III-F plate by means of the Gaertner medium quartz spectrograph. The amount of manganese was determined from the density of the line at 2798.27 A. U. compared with the densities in spectra of standard samples exposed on the same plate. The standard samples consisted of reagent grade sodium chloride (Mn free) containing graduated amounts of manganous chloride. The analytical results were as follows:
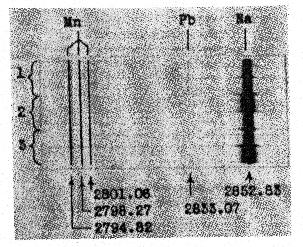
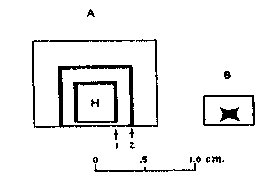
The Idaho samples fluoresce more weakly than do the samples from Amboy, California, but, as shall be shown later, the brightness of fluorescence is not determined entirely by the concentration of manganese. CHEMICAL OBSERVATIONS When the fluorescing halite specimens were dissolved in water, and the solution filtered and evaporated, the resulting crystals also fluoresced red. This showed that the activator was either soluble in water or, as seems less probable, was of colloidal size. Both the natural and synthetic specimens turned a faint brown and lost their fluorescence when heated for a few hours on a hot plate at around 300°C. When such heated crystal were recrystallized in water, they did not regain their fluorescence but would do so if a few drops of hydrochloric acid were added to the water before recrystallizing. Reagent grade sodium chloride was carried through these tests as a blank. It did not turn brown on heating or show a fluorescence at any stage of the experiment. These various effects may be accounted for satisfactorily on the basis that bivalent manganese is essential for producing the fluorescence. The development of a brown color and the loss of fluorescence upon heating are due to the bivalent manganese being oxidized to one of the higher oxides of manganese through reaction with atmospheric oxygen. The restoration of fluorescence through the use of hydrochloric acid may be explained by the reducing action that this acid has on the higher manganese oxides which converts manganese back to the bivalent state Organic compounds, which are known to cause fluorescence in some minerals, seem to be definitely eliminated from consideration by the tests described above. Iron or cobalt might also show the effects that were observed. Iron was readily eliminated by a trial synthesis that showed that it produces no fluorescence in salt. The result for cobalt was also negative though less decisive. A very faint reddish fluorescence was seen in crystals grown from a cobaltiferous mother liquor, but the "chemically pure" cobaltous chloride that was used in the experiment was later found to contain a small amount of manganese. This illustrates the difficulty caused by impurities that are present in reagents in amounts that are negligible for ordinary chemical purposes but loom large where fluorescence effects are involved. Cobalt was not found in any of the many samples of both natural and synthetic fluorescent halite which were analyzed spectrographically during the course of this study. Thus all evidence points to small amounts of manganese activating a red fluorescence in sodium chloride, and the story would be complete were it not for a puzzling localization of fluorescence that was noted in both the natural and synthetic specimens. LOCALIZATION OF FLUORESCENCE The crystalline aggregates that make up the samples from the three Western localities do not give off a uniformly diffused fluorescence, but rather show small areas, and in some specimens, lateral zones which fluoresce brighter than the rest. This is best seen in the more coarsely crystallized samples that come from the two California localities. FIG. 2. Localized fluorescence in synthetic manganiferous salt crystals. A. Vertical section of a crystal grown with a cleavage piece of nonfluorescing halite (H) as a seed nucleus. Fluorescent zone 1 deposited during the first day from manganiferous brine. Nonfluorescent portion between 1 and 2 deposited during the next three days. Fluorescent zone 2 deposited upon addition of small amount of lead nitrate to mother liquor. B. Side view of a crystal with typical, hopper-shaped fluorescent core obtained from unseeded manganiferous brine. In growing salt crystals in the laboratory, it was noted that invariably only the crystals that were deposited during the first day or so from manganiferous brine fluoresced brightly, and later crops were either only feebly fluorescent or not at all. Moreover, with the crystals of the first crop the fluorescence was restricted to that part of the crystal that formed first, namely, the hopper shaped core shown in Fig. 2 B. When pieces of natural halite selected for their uniform fluorescence were dissolved in water and crystallized, again only the first formed crystal fluoresced, and these had their fluorescence restricted to the core as shown in Fig. 2 B. The same phenomenon was seen when a cleavage piece of non-fluorescent halite from Detroit, Michigan, was used as a seed (see Fig. 2 A). Only a narrow zone, marked No. 1, nearest the original seed fluoresced brightly and the crystal continued to grow by accretion of nonfluorescent salt. Samples from the two California localities contain many crystals with just such narrow fluorescent zones. All laboratory syntheses were carried out with 33.5 ml. portions of a stock solution of reagent grade sodium chloride saturated at room temperature (25°C.). Reagent grade MnCl 2·4H2O was weighed and added to the saturated brine, which was then undersaturated with four drops of water and allowed to evaporate in a 100 ml. pyrex beaker. Amounts of manganese less than a milligram were introduced into the saturated brine by adding the required number of calibrated drops of a manganous chloride solution of known strength. When successive crops of salt were harvested, the mother liquor was decanted into a 50 ml. beaker every two days and undersaturated with three drops of water before further evaporation. The range of initial concentrations of manganese studied was 0.083 to 1,200 mg. Mn per liter.The deposition of nonfluorescent salt after the first day or so in the various experiments was puzzling because the mother liquor stilt contained most of the manganese which was initially added. The mother liquor of one preparation was decanted off after it had deposited nonfluorescent salt for several days and the manganese in it was determined gravimetrically as the pyrophosphate. Eighty per cent of the original manganese was present in half the original volume. Yamamoto, 12 who studied the influence of various metal ions on the growth of alkali salt crystals from water solutions, has shown that over a considerable range of manganese concentration the amount of manganese in the first crop of salt may be represented as an exponential function of the manganese concentration in solution, and in general is only a few per cent of what it would be if manganese and salt were deposited in the ratio prevailing in the mother liquor. Thus there would be an increase in the concentration of dissolved manganese, as the deposition proceeded and the volume of solution decreased through evaporation.In preparing thallium-activated, blue-fluorescing potassium chloride from water solutions, Pringsheim 13 noted that the early crops fluoresced most brightly, but here the explanation was found in the rapid coprecipitation of thallium by potassium chloride. For example, Pringsheim found that by the time 5% of the potassium chloride in a saturated solution had deposited, 99% of the thallium originally present had been removed from solution.The simplest explanation that occurred to us for the localization of fluorescence in our salt crystals was that some unexpected impurity was present in our solutions, that this impurity was also necessary for producing the red fluorescence, and that it was rapidly removed from solution by the first crops of salt just as thallium is by growing crystals of potassium chloride. This explanation proved to be correct, and lead was identified as the second essential activating element. The presence of traces of lead in all of our natural and synthetic fluorescent salt samples had been established spectrographically early in the study, but the element was not given serious consideration at first for several reasons, (1) its amount was small, (2) it was not known to activate fluorescence in very many compounds, and finally, (3) traces of lead had been encountered in so many rocks and minerals and in practically all chemical reagents during the course of spectrographic work in our laboratory that we had fallen into the habit of accepting traces of lead as commonplace and uninteresting. The clue that finally pointed to lead as the second essential activating element was found in some spectrograms taken to test the unlikely possibility that, for some reason, growing salt crystals stopped adsorbing manganese after the first couple of days, and that later crystals would not fluoresce because they contained no manganese. Three successive, two-day crops from one manganiferous mother liquor were spectrographically analyzed in duplicate, the first crop being, as usual, the only one that fluoresced. Figure 3 shows a small portion of duplicate spectra of these three crops in the region of 2800 A. U., and the three sensitive lines of manganese (2794.82, 2798.27, and 2801.06 A. U.) are seen to be of same strength in all of the spectra, indicating that the nonfluorescent crystals of crops 2 and 3 contain as much manganese as do the fluorescent crystals of crop 1. Fortunately, the most sensitive line of lead is also located in this spectral region, and lead, which previously had been ignored, forced itself upon our attention. The minute amount of lead which had been present in our reagents as a ubiquitous impurity was shown concentrated in the only crop of crystals that fluoresced. With this clue, a small amount of lead nitrate was purposely added to a manganiferous brine that had been depositing nonfluorescent salt around a seed crystal for several days, and to our delight another fluorescent zone formed like the one marked No. 2 in Fig. 2 A. By varying the amount of lead, fluorescent zones of any thickness could be formed around a seed crystal, or a mother liquor could be made at will to deposit crops of fluorescent crystals for any number of days.
FIG. 3. Spectrographic analysis, in duplicate, of three successive crops of salt from a manganiferous mother liquor. The lead impurity is seen concentrated in the first crop, the only one that fluoresced. Wavelengths in angstrom units. Neither manganese nor lead alone will produce a fluorescence or, at most, only a barely detectable fluorescence at high concentrations in the salt. But the two together in amounts of the order of a few thousandths of a per cent will coactivate a vivid red fluorescence. ADSORPTION OF MANGANESE Because manganese, unlike lead, is not removed in disproportionate amounts from solution by growing crystals of salt and because reagent grade sodium chloride is essentially free of manganese, it was possible to determine a lower limit of concentration of manganese below which the first crop of crystals did not fluoresce even when an adequate amount of lead was present in the mother liquor. A series of saturated salt solutions were made up to contain a rather high concentration of lead (1 mg. Pb per liter) with variable amounts of manganese, and the fluorescence of the first crops was noted. The results are assembled in Table 1. With the mother liquor containing 0.08 mg. Mn per liter, a barely detectable fluorescence of uncertain hue was produced when the initial lead concentration was raised to 20 mg. Pb per liter, a value probably never attained in nature. This feeble fluorescence is probably due largely to lead as a blank preparation (No. 7), also high in lead, produced crystals that fluoresced similarly. Crystals of preparation No. 4 fluoresced more weakly than do the samples from the two California desert localities. The conclusion seems to be indicated that the desert brine at these localities must have contained at least several milligrams manganese per liter and probably even greater amounts, if, as was likely, the lead concentration was smaller than 1 mg. Pb per liter. TABLE 1. COMPOSITION OF MOTHER LIQUORS AND FLUORESCENCE OF THEIR FIRST CROPS OF CRYSTALS
Natural bottom waters, both fresh and marine, are known to contain as much as 15 mg. Mn per liter, when they lie stagnant over bottom sediments in which anaerobic decomposition of organic matter is proceeding actively. Concentrations as high as 50 mg. Mn per liter were noted by Robinson 14 in laboratory experiments with submerged soils. However, if fluorescent salt crystals are deposited from a brine of such a chemically reducing nature the brine would have to be low in sulfate; otherwise, hydrogen sulfide may be generated which would precipitate lead sulfide and reduce the concentration of dissolved lead. Manganese may also be precipitated as the sulfide, but the acidity resulting from carbonic acid liberated by decomposing organic matter would tend to keep the element in solution.The crystals of preparation No. 1, whose mother liquor contained 400 mg. Mn per liter, were found spectrographically to contain 0.008% Mn. For our purpose it may be safely assumed that in the lower ranges the amount in the deposited salt is directly proportional to the concentration in solution, so a mother liquor containing 40 mg. Mn per liter would be expected to yield salt with a manganese content of around 0.0008%, a value well within the range found for natural, fluorescent halite. COPRECIPITATI0N OF LEAD The abstraction of disproportionately large amounts of lead from brines by growing sodium chloride crystals has been demonstrated by Kading, 15 who worked with extremely low concentrations of the order of 3 X 10-6g mg. Pb per liter; and by Yamamoto,16 who covered the range of 21-561 mg. Pb per liter. In our use of lead nitrate to synthesize fluorescent salt, we made up the manganiferous mother liquors to contain initial concentrations of 0.9-93.0 mg. Pb per liter.For the concentration range covered in our respective work, both Yamamoto and we found it necessary to use small amounts of either nitric or hydrochloric acid to prevent the lead from precipitating as the chloride. Our early syntheses were carried out with solutions to which no lead had been added, but which nevertheless contained it as an unsuspected impurity, in an amount roughly estimated to be around 0.1 mg. Pb per liter, largely contributed by the reagent-grade sodium chloride. No acid was added to these solutions, but the same rapid removal of lead was observed with them as with the solutions holding larger amounts of lead. The early removal of most of the dissolved lead by growing salt crystals thus would tend to limit the quantity of fluorescent salt that a given manganiferous brine could produce, and would also cause localisation of fluorescence within crystals when the supply of lead is limited. Although the removal of lead is rapid in the beginning, the rate becomes slower with decreasing concentration. Small amounts persist in solution and appear as spectrographic traces in the later crops of crystals This probably explains the persistence of a feeble red fluorescence in later crops of crystals from mother liquors containing high concentrations of manganese of the order of 1000-2000 mg. Mn per liter. It is evident that the brightness of fluorescence depends on the amounts of both lead and manganese, and within limits a higher content of one will compensate for a deficiency of the other. Nevertheless, as with manganese, there appears to be a limiting value of lead below which a bright fluorescence is not produced no matter how great an amount of manganese is present in the crystal. Fluorescent zones like those shown in Fig. 2 show fairly sharp outer boundaries, instead of gradual transition from bright to dim fluorescence, which also suggests the reality of such a limiting concentration of lead. The lead content of the natural, brightly fluorescent halite specimens has not been accurately determined, but a rough estimate based on the intensity of the 2833.07 A. U. line in the spectrograms would be 0.001-.01% Pb. MISCELLANEOUS OBSERVATIONS As barium, univalent thallium, and bismuth are somewhat similar in their chemical behavior to lead, small amounts of their salts were tried as substitutes for lead in coactivating the red fluorescence with manganese, but the results were negative. Stannous chloride with manganese also gave a negative result. As far as can be told from our brief survey, the manganese-lead pair is unique in producing the red fluorescence; and the relationship may find use as a basis for a qualitative test for small amounts of lead. A manganiferous, saturated salt solution undergoing evaporation is extremely sensitive to lead; and will betray even very small amounts of it by depositing fluorescent salt. This was illustrated in one preparation in which an acid-free mother-liquor, which had become depleted of its lead, "crept" up the side of the beaker and coated the outside with a thin layer of salt. Under the ultraviolet lamp the Pyrex brand on the beaker fluoresced bright red against a background of nonfluorescent salt. By itself, the brand does not fluoresce at all. Subsequently, it was learned that a flux of lead borate is used to fix the brand on the glass surface. The creeping salt solution had extracted enough lead from the letters to cause the salt deposited over them to fluoresce brightly. The aim in our study has been to work out the gross, qualitative aspects of this interesting coactivation of a red fluorescence in sodium chloride by manganese and lead, and it is evident that the system deserves a more thorough and precise study. ACKNOWLEDGMENTS We are indebted to Mr. Thomas S. Warren of Los Angeles, California, for samples of red-fluorescing halite from the two California localities. Drs. W. T. Schaller, C. S. Ross, Michael Fleischer, and Mr. W. G. Schlecht of the Geological Survey have critically read the manuscript of the paper and made many helpful suggestions. We express our hearty thanks to Mr. Warren and our colleagues for their generous help.
NOTES * Published by permission of the Director, Geological Survey, U. S. Department of the Interior, Washington, D. C. ** On military leave. 1 The fluorescence color of the natural and synthetic halite specimens examined during the course of our study included various tints of red. Most specimens fluoresced a deep pink. However, the term red seems preferable to pink because red has been used in all previous descriptions of this fluorescence in the literature.
2 Brown, W. L., Univ. Toronto Studies, Geol. Ser. no. 36, 45-54 (1934). 3 Przibram, Karl, Akad. Wiss.Wien, math.-naturwiss. Kl., Sitzungsber.,Abt. IIa, 134, 234-235 (1925). 4 Jahoda, Eduard, Idem, 135, 675-703 (197.6). 5 Oka, Sojiro, and Yagi, Sakaye, Jour. Soc. Chem. Ind. Japan, Supplem. Binding, 36, 143B-144B (1933). 6 Randall, J. T., Proc. Roy. Soc. London, ser. A,170, 272-293 (1939). 7 Dake, H. C., and DeMent, Jack, Ultraviolet Light and Its Applications, p.129. Chemical Publishing Co., Brooklyn (1941). 8 Personal communication from Thomas S. Warren of Los Angeles, California, April 14, 1944. We are indebted to Mr. Warren for a number of specimens from the two California localities. 9 Phalen, W. C., U. S. Geol. Survey, Bull. 669, 132 (1919). 10 Phalen, W. C., op. cit., p. 185. 11 Mansfield, G. R, U. S. Geol. Survey, Prof. Paper, 152, 340 (1927). 12 Yamamoto, Takemaro, Sci. Papers Inst. Phys. Chem. Research, 35, 228-289 (1939). 13 Pringsheim, Peter, Rev. Mod. Physics, 14, 132-138 (1942). 14 Robinson, W. O., Soil Sci., 30, 197-217 (1930). 15 Kading, Hans, Zeit. physik. Chem., 362A, 174-186 (1932). See also Hahn, Otto, Applied Radiochemistry, pp. 102113, Cornell Univ. Press (1936). 16 Yamamoto, Takemaro, op, cit., pp. 264-262.
| |||||||||||||||||||||||||||||||||||||||||||||||||||